

Deep learning is a type of artificial intelligence in which computers learn to identify and categorize patterns in huge data sets. A start-up, Deep Genomics, that thinks it can transform medicine by applying the hot field of deep learning to genomics.
Brendan Frey and his team would spin out of the University of Toronto and launch a start-up —Deep Genomics.
Frey had gradually realized how rare his expertise in both deep learning and genetics was. Deep learning is currently a hot field in technology, but it’s a small one.
Much of the talent has passed through the University of Toronto’s deep learning group. For example, Yann LeCun now leads Facebook’s artificial intelligence research lab. Geoffrey Hinton, who Frey had worked on machine vision with, now works at Google. Yoshua Bengio is collaborating with IBM’s Watson group.
Deep Genomics will try to tap into the 98.5 percent of the genome that hasn’t been studied closely for mutations and looks for the disease consequences of any mutations it finds.
The idea is to lay the foundation for computers to one day be in charge of predicting lab experiments and treatments and to guide drug development and personalized medicine – perhaps eventually reaching as low as your local doctor’s office. This is really just the beginning. Deep Genomics technology is already finding commercial use in medical diagnostics and drug development.
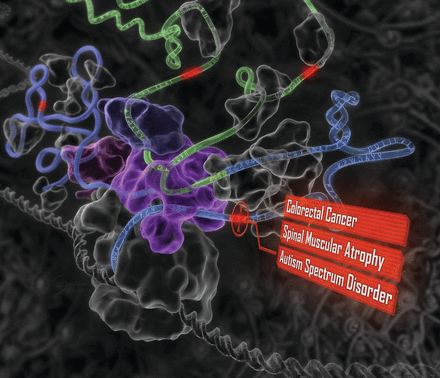
A computational system mimics the biology of RNA splicing by correlating DNA elements with splicing levels in healthy human tissues. The system can scan DNA and identify damaging genetic variants, including those deep within introns. This procedure has led to insights into the genetics of autism, cancers, and spinal muscular atrophy.
Science – The human splicing code reveals new insights into the genetic determinants of disease
Predicting defects in RNA splicing
Most eukaryotic messenger RNAs (mRNAs) are spliced to remove introns. Splicing generates uninterrupted open reading frames that can be translated into proteins. Splicing is often highly regulated, generating alternative spliced forms that code for variant proteins in different tissues. RNA-binding proteins that bind specific sequences in the mRNA regulate splicing. Xiong et al. develop a computational model that predicts splicing regulation for any mRNA sequence (see the Perspective by Guigó and Valcárcel). They use this to analyze more than half a million mRNA splicing sequence variants in the human genome. They are able to identify thousands of known disease-causing mutations, as well as many new disease candidates, including 17 new autism-linked genes.
INTRODUCTION
Advancing whole-genome precision medicine requires understanding how gene expression is altered by genetic variants, especially those that are far outside of protein-coding regions. We developed a computational technique that scores how strongly genetic variants affect RNA splicing, a critical step in gene expression whose disruption contributes to many diseases, including cancers and neurological disorders. A genome-wide analysis reveals tens of thousands of variants that alter splicing and are enriched with a wide range of known diseases. Our results provide insight into the genetic basis of spinal muscular atrophy, hereditary nonpolyposis colorectal cancer, and autism spectrum disorder.RATIONALE
We used “deep learning” computer algorithms to derive a computational model that takes as input DNA sequences and applies general rules to predict splicing in human tissues. Given a test variant, which may be up to 300 nucleotides into an intron, our model can be used to compute a score for how much the variant alters splicing. The model is not biased by existing disease annotations or population data and was derived in such a way that it can be used to study diverse diseases and disorders and to determine the consequences of common, rare, and even spontaneous variants.RESULTS
Our technique is able to accurately classify disease-causing variants and provides insights into the role of aberrant splicing in disease. We scored more than 650,000 DNA variants and found that disease-causing variants have higher scores than common variants and even those associated with disease in genome-wide association studies (GWAS). Our model predicts substantial and unexpected aberrant splicing due to variants within introns and exons, including those far from the splice site. For example, among intronic variants that are more than 30 nucleotides away from any splice site, known disease variants alter splicing nine times as often as common variants; among missense exonic disease variants, those that least affect protein function are more than five times as likely as other variants to alter splicing.Autism has been associated with disrupted splicing in brain regions, so we used our method to score variants detected using whole-genome sequencing data from individuals with and without autism. Genes with high-scoring variants include many that have previously been linked with autism, as well as new genes with known neurodevelopmental phenotypes. Most of the high-scoring variants are intronic and cannot be detected by exome analysis techniques.
When we scored clinical variants in spinal muscular atrophy and colorectal cancer genes, up to 94% of variants found to alter splicing using minigene reporters were correctly classified.
CONCLUSION
In the context of precision medicine, causal support for variants independent of existing whole-genome variant studies is greatly needed. Our computational model was trained to predict splicing from DNA sequence alone, without using disease annotations or population data. Consequently, its predictions are independent of and complementary to population data, GWAS, expression-based quantitative trait loci (QTL), and functional annotations of the genome. As such, our technique greatly expands the opportunities for understanding the genetic determinants of disease.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.