Pancreatic cancer carries a very bleak prognosis for patients. However, a recent breakthrough by two research teams, including one at the Hôpital Maisonneuve-Rosemont (CIUSSS-EST, Montreal) and University of Montreal, has opened the door to a better understanding of the molecular mechanisms that cause this cancer to develop.
This biomedical research conducted jointly by the groups of Dr. Frédérick Antoine Mallette (Université de Montréal / Centre de Recherche HMR) and Dr. Stéphane Richard (McGill University / Lady Davis Institute for Medical Research) and that was published in Cell Reports has shown that pancreatic tumors often lose the ability to express a small ribonucleic acid molecule called miR-137. This molecule induces a defence mechanism called cellular senescence, which keeps cancer cells in check. The loss of miR-137 works in conjunction with various mutations frequently observed in pancreatic tumours to trigger uncontrolled cell growth and then cancer.
This joint research study by doctoral student Mathieu Neault has also demonstrated that restoring normal miR-137 levels in pancreatic cancer cells has a protective effect, as doing so induces senescence and stops the cells from spreading.
In 2015, approximately 4800 people received a diagnosis of pancreatic cancer, and nearly 4600 Canadians succumbed to this terrifying disease. Although this cancer is the 12th highest in terms of incidence, it is 4th highest in cancer-related mortality. Survival rates for pancreatic cancer haven’t improved in the past 40 years. This is why we urgently need to clarify the mechanisms of this cancer to find new therapeutic avenues that will change these grim statistics.

Highlights
•miR-137 triggers the p53 and p16INK4A tumor suppressor pathways
•KDM4A is a target of miR-137 during Ras-induced senescence
•Loss of miR-137 contributes to the bypass of Ras-induced senescence
•Restoration of miR-137 expression induces senescence in pancreatic cancer cells
Summary
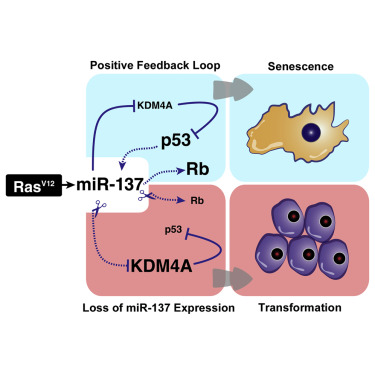
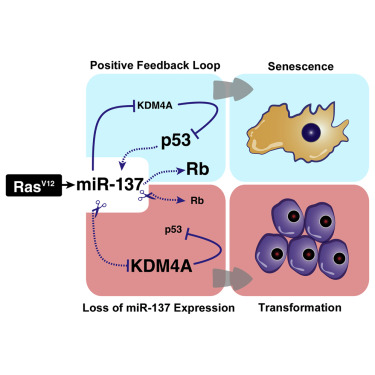
Activating K-Ras mutations occurs frequently in pancreatic cancers and is implicated in their development. Cancer-initiating events, such as oncogenic Ras activation, lead to the induction of cellular senescence, a tumor suppressor response. During senescence, the decreased levels of KDM4A lysine demethylase contribute to p53 activation, however, the mechanism by which KDM4A is downregulated is unknown. We show that miR-137 targets KDM4A mRNA during Ras-induced senescence and activates both p53 and retinoblastoma (pRb) tumor suppressor pathways. Restoring the KDM4A expression contributed to bypass of miR-137-induced senescence and inhibition of endogenous miR-137 with an miRNA sponge-compromised Ras-induced senescence. miR-137 levels are significantly reduced in human pancreatic tumors, consistent with previous studies revealing a defective senescence response in this cancer type. Restoration of miR-137 expression inhibited proliferation and promoted senescence of pancreatic cancer cells. These results suggest that modulating levels of miR-137 may be important for triggering tumor suppressor networks in pancreatic cancer.
In this paper, we describe the ability of miR-137 to induce cellular senescence. miR-137 levels are increased during OIS and in response to DNA damage. Ectopic expression of miR-137 led to a cell-cycle arrest phenotype associated with the hallmark of premature cellular senescence. We demonstrate that miR-137 engages both ARF/p53 and p16INK4A/pRb pathways to promote cellular senescence, and we have identified the mRNA of the KDM4A lysine demethylase as an miR-137 target. KDM4A previously was shown to decrease during OIS and its restoration allowed bypass of cellular senescence (Mallette and Richard, 2012). We now show that downregulation of KDM4A by miR-137 activates the CHD5/p53 tumor suppressor pathway. In addition, expression of an miRNA-insensitive KDM4A partially rescued miR-137-induced senescence, while a complete rescue was achieved with the concomitant expression of KDM4A and depletion of p16INK4A, consistent with miR-137 engaging both ARF/p53 and p16INK4A/pRb pathways. Inhibition of endogenous miR-137 using an miRNA sponge in IMR90 severely compromised the OIS response and engendered a significant decrease of CDKN1A expression, thus confirming the contribution of miR-137 in the activation of p53 and establishment of cellular senescence. In human pancreatic tumors, we show that the expression of three transcriptional targets repressed by KDM4A, CHD5, ASCL2, and PANX2, correlated with miR-137 expression levels. This observation suggests that miR-137, by decreasing KDM4A levels, relieved KDM4A-mediated repression on these genes. Ectopic expression of miR-137 in pancreatic cancer cell line PANC-1 halted cell proliferation and induced cellular senescence, a cellular response also observed upon the depletion of KDM4A. These findings suggest that increasing miR-137 in pancreatic cancer might be of therapeutic significance.
Reduced expression levels of mature miR-137 has been reported in many tumor types including glioblastoma (Chen et al., 2012) and ovarian cancer (Guo et al., 2013), and also Ras-driven colorectal cancer (Chen et al., 2013), non-small cell lung carcinoma (Zhang et al., 2015), and prostate cancer (Nilsson et al., 2015). Furthermore, miR-137 was shown to reduce invasion while increasing sensitivity to chemotherapy in pancreatic cancer (Xiao et al., 2014). We now define the molecular mechanism of action of miR-137. In this respect, we have defined herein the molecular underpinnings by showing that miR-137 stimulates a tumor suppressor network engaging both ARF/p53 and p16INK4A/pRb pathways to promote cellular senescence in pancreatic cancer cells. Taken together, our data also suggest that the ability of miR-137 to induce senescence may be relevant to many cancer types.
Activation of the DNA damage response is known to stimulate the maturation of specific miRNAs (Chowdhury et al., 2013). We show that the expression of miR-137 is induced by DNA damage, and further experimentation is required to define whether miR-137 is a direct target of p53. Attempts to identify chromatin-bound p53 at putative p53 response elements in the miR-137 upstream region by ChIP were unsuccessful (data not shown). It is possible that we have not identified the appropriate p53-binding site. p53 has been shown to stimulate the maturation of numerous miRNAs (Suzuki et al., 2009), and this also may be a mechanism by which miR-137 is regulated by p53. Thus, we have uncovered a mechanism whereby miR-137 and p53 promote each other’s activation in a positive feedback loop during the DNA damage response and following oncogenic stress).
PDAC is the most frequent form of pancreatic cancer (∼90% of the cases), is highly aggressive, and exhibits a very poor 5-year survival rate (less than5%) (Bryant et al., 2014, Eser et al., 2014). Activating K-Ras mutations is recognized as a primary determinant in the initiation of pancreatic intraepithelial neoplasia (PanIN) (Eser et al., 2014), however, progression from low-grade PanIN to PDAC often requires the chronological disruption of the INK4A/ARF tumor suppressor locus followed by p53 inactivation (Hezel et al., 2006), leading to genomic instability and tumor progression (Bryant et al., 2014, Hezel et al., 2006). miR-137 expression is decreased in pancreatic tumors, and its restoration in PANC-1 cells induced cellular senescence response and growth inhibition. Accordingly, downstream miR-137 signaling seems intact and functional in pancreatic cancers. Additionally, bypass of OIS observed upon miR-137 loss of function greatly supports the notion that the loss of miR-137 is an early event driving pancreatic tumorigenesis in cooperation with activated Ras.
We observed that miR-137 levels are lower in a pancreatic tumor tissue cohort versus normal tissues. miR-137 expression correlated with levels of KDM4A downstream negative targets CHD5, ASCL2, and PANX2 and also with KDM4A positively modulated target PPIC. The levels of mRNAs for KDM4A in these pancreatic cancer samples did not correlate with decreased miR-137 levels. Although miR-137 might primarily regulate KDM4A mRNA translation rather than mRNA degradation, it is possible that there are other factors that preclude us from observing KDM4A mRNA decay, such as elevated levels of steady-state mRNAs. Moreover, we observed that the half-life of the KDM4A mRNA in cycling fibroblasts is similar to senescent fibroblasts induced by RasV12. This may be explained by the fact that miRNA-mediated silencing is not solely the result of mRNA decay, but also can be attributed to 5′ cap-dependent translation inhibition (Filipowicz and Sonenberg, 2015). Thus, the transcriptional analysis of KDM4A downstream targets constituted a more informative measure of KDM4A activity than KDM4A mRNA levels.
It is important to consider endogenous miRNA expression levels when studying miRNA-mediated translation inhibition. miR-137 is annotated with 54 reads per million in the latest version of miRBase database (http://www.mirbase.org/). Other low-copy miRNAs, such as the miR-34 family (e.g., miR-34b annotated with 294 reads on miRBase), have been shown to induce senescence, and their loss of expression is a common feature in cancer (Hermeking, 2010).
Despite relatively low amounts of miR-137, its >5-fold increase observed in our RasV12-induced senescence model (Figure S2A) reaches the same levels as miR-34. In addition, inhibition of miR-137 endogenous activity significantly impaired the OIS response (Figure 4), and its expression was undetected in pancreatic cancer cells. These results are consistent with our findings that miR-137 is lost in pancreatic cancer, and they reveal that miR-137 is a physiologically relevant mediator of OIS.
miR-137 stimulates cellular senescence by engaging the p16INK4A/pRb and the ARF/p53 pathways independently (Figures 2C and 3A). The partial contribution of KDM4A in rescuing miR-137-induced senescence (Figure 2D) implies that there are other effectors of miR-137 involved in promoting senescence. miR-137 is known to target cyclin-dependent kinase CDK6 and the Rho-GTPase family member CDC42 (Liu et al., 2011, Zhu et al., 2013). CDK6 forms a complex with cyclin D to phosphorylate pRb and release it from E2F-dependent genes, thus permitting S phase entry (Choi and Anders, 2014). CDC42 controls cell-cycle progression by stimulating cyclin D expression (Welsh et al., 2001), and it cooperates with oncogenic Ras during transformation (Qiu et al., 1997). Thus, miR-137 might regulate the p16INK4A/pRb pathway at multiple levels, possibly through downregulation of CDK6 and CDC42. On the other hand, miR-137 dampens KDM4A activity to stimulate the p53 pathway in a positive feedback loop and also triggers the p16INK4A/pRb pathway, which leads to cell-cycle arrest and senescence (Figure 7). KDM4A mRNA is a main target of miR-137 in the ARF/p53 pathway, since the concomitant restoration of miR-137-resistant KDM4A in p16INK4A-depleted cells leads to a complete bypass of senescence.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.