

Google has performed the first completely scalable quantum simulation of a molecule. Google worked with researchers from the Aspuru-Guzik group at Harvard and researchers from Lawrence Berkeley National Labs, UC Santa Barbara, Tufts University and University College London.
The goal of their experiment was to use quantum hardware to efficiently solve the molecular electronic structure problem, which seeks the solution for the lowest energy configuration of electrons in the presence of a given nuclear configuration. In order to predict chemical reaction rates (which govern the mechanism of chemical reactions), one must make these calculations to extremely high precision. The ability to predict such rates could revolutionize the design of solar cells, industrial catalysts, batteries, flexible electronics, medicines, materials and more. The primary difficulty is that molecular systems form highly entangled quantum superposition states which require exponentially many classical computing resources in order to represent to sufficiently high precision. For example, exactly computing the energies of methane (CH4) takes about one second, but the same calculation takes about ten minutes for ethane (C2H6) and about ten days for propane (C3H8).
In their experiment, they focus on an approach known as the variational quantum eigensolver (VQE), which can be understood as a quantum analog of a neural network. Whereas a classical neural network is a parameterized mapping that one trains in order to model classical data, VQE is a parameterized mapping (e.g. a quantum circuit) that one trains in order to model quantum data (e.g. a molecular wavefunction). The training objective for VQE is the molecular energy function, which is always minimized by the true ground state. The quantum advantage of VQE is that quantum bits can efficiently represent the molecular wavefunction whereas exponentially many classical bits would be required

Physical Review X – Scalable Quantum Simulation of Molecular Energies
Using VQE, they quantum computed the energy landscape of molecular hydrogen, H2. They compared the performance of VQE to another quantum algorithm for chemistry, the phase estimation algorithm (PEA). Experimentally computed energies, as a function of the H – H bond length, are shown below alongside the exact curve. They were able to obtain such high performance with VQE because the neural-network-like training loop helped to establish experimentally optimal circuit parameters for representing the wavefunction in the presence of systematic control errors. One can understand this by considering a hardware implementation of a neural network with a faulty weight, e.g. the weight is only represented half as strong as it should be. Because the weights of the neural network are established via a closed-loop training procedure which can compensate for such systematic errors, the hardware neural network is robust against such imperfections. Likewise, despite systematic errors in our implementation of the VQE circuit, we are still able to learn an accurate model for the wavefunction. This robustness inspires hope that VQE may be able to solve classically intractable problems without quantum error correction.
While the energies of molecular hydrogen can be computed classically (albeit inefficiently), as one scales up quantum hardware it becomes possible to simulate even larger chemical systems, including classically intractable ones. For instance, with only about a hundred reliable quantum bits one could model the process by which bacteria produce fertilizer at room temperature. Elucidating this mechanism is a famous open problem in chemistry because the way humans produce fertilizer is extremely inefficient and consumes 1-2% of the world’s energy annually. Such calculations could also assist with breakthroughs in fundamental science, for instance, in the understanding of high temperature superconductivity.
Though many theoretical and experimental challenges lay ahead, a quantum enabled paradigm shift from qualitative / descriptive chemistry simulations to quantitative / predictive chemistry simulations could modernize the field so dramatically that the examples imaginable today are just the tip of the iceberg.
Abstract
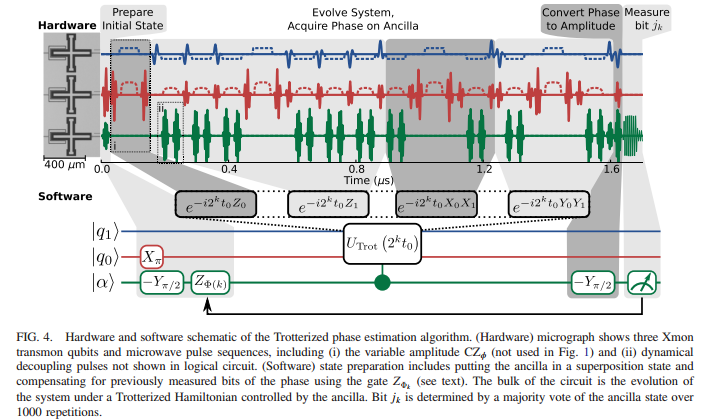
We report the first electronic structure calculation performed on a quantum computer without exponentially costly precompilation. We use a programmable array of superconducting qubits to compute the energy surface of molecular hydrogen using two distinct quantum algorithms. First, we experimentally execute the unitary coupled cluster method using the variational quantum eigensolver. Our efficient implementation predicts the correct dissociation energy to within chemical accuracy of the numerically exact result. Second, we experimentally demonstrate the canonical quantum algorithm for chemistry, which consists of Trotterization and quantum phase estimation. We compare the experimental performance of these approaches to show clear evidence that the variational quantum eigensolver is robust to certain errors. This error tolerance inspires hope that variational quantum simulations of classically intractable molecules may be viable in the near future.
SOURCES- Google Blog, Physical Review X

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.