

Yale School of Medicine researchers have discovered a protein that is the missing link in the complicated chain of events that lead to Alzheimer’s disease, they report in the Sept. 4 issue of the journal Neuron. Researchers also found that blocking the protein with an existing drug can restore memory in mice with brain damage that mimics the disease.
In the Neuron paper, the Yale team reveals the missing link in the chain, a protein within the cell membrane called metabotropic glutamate receptor 5 or mGluR5. When the protein is blocked by a drug similar to one being developed for Fragile X syndrome, the deficits in memory, learning, and synapse density were restored in a mouse model of Alzheimer’s.
Strittmatter stressed that new drugs may have to be designed to precisely target the amyloid-prion disruption of mGluR5 in human cases of Alzheimer’s and said his lab is exploring new ways to achieve this.
Highlights
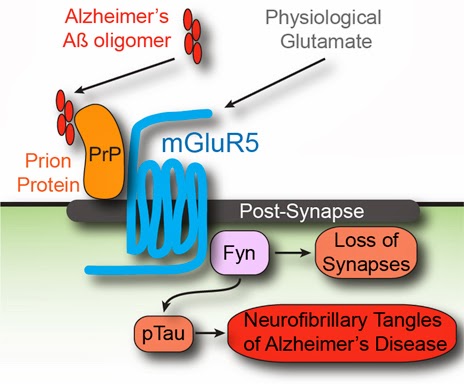
• Among transmembrane PSD proteins, only mGluR5 couples Aβo-PrPC to Fyn kinase
• mGluR5 also links Aβo-PrPC to calcium signaling and protein translation control
• AD brain extract-induced dysregulation of neuronal calcium requires PrPC-mGluR5
• Transgenic mouse memory deficits and synapse loss are reversed by mGluR5 antagonist
Summary
Soluble amyloid-β oligomers (Aβo) trigger Alzheimer’s disease (AD) pathophysiology and bind with high affinity to cellular prion protein (PrPC). At the postsynaptic density (PSD), extracellular Aβo bound to lipid-anchored PrPC activates intracellular Fyn kinase to disrupt synapses. Here, we screened transmembrane PSD proteins heterologously for the ability to couple Aβo-PrPC with Fyn. Only coexpression of the metabotropic glutamate receptor, mGluR5, allowed PrPC-bound Aβo to activate Fyn. PrPC and mGluR5 interact physically, and cytoplasmic Fyn forms a complex with mGluR5. Aβo-PrPC generates mGluR5-mediated increases of intracellular calcium in Xenopus oocytes and in neurons, and the latter is also driven by human AD brain extracts. In addition, signaling by Aβo-PrPC-mGluR5 complexes mediates eEF2 phosphorylation and dendritic spine loss. For mice expressing familial AD transgenes, mGluR5 antagonism reverses deficits in learning, memory, and synapse density. Thus, Aβo-PrPC complexes at the neuronal surface activate mGluR5 to disrupt neuronal function.
If you liked this article, please give it a quick review on ycombinator or StumbleUpon. Thanks

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.