

Researchers have discovered a cause of aging in mammals that may be reversible.
UPDATE – Correcting to the right Harvard article for the new 2013 research.
The essence of this finding is a series of molecular events that enable communication inside cells between the nucleus and mitochondria. As communication breaks down, aging accelerates. By administering a molecule naturally produced by the human body, scientists restored the communication network in older mice. Subsequent tissue samples showed key biological hallmarks that were comparable to those of much younger animals.
Highlights
• A specific decline in mitochondrially encoded genes occurs during aging in muscle
• Nuclear NAD+ levels regulate mitochondrial homeostasis independently of PGC-1α/β
• Declining NAD+ during aging causes pseudohypoxia, which disrupts OXPHOS function
• Raising nuclear NAD+ in old mice reverses pseudohypoxia and metabolic dysfunction
Summary
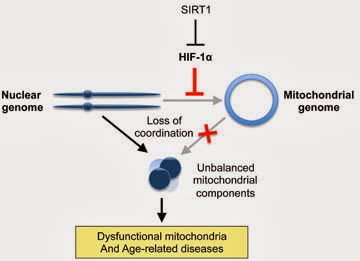
Ever since eukaryotes subsumed the bacterial ancestor of mitochondria, the nuclear and mitochondrial genomes have had to closely coordinate their activities, as each encode different subunits of the oxidative phosphorylation (OXPHOS) system. Mitochondrial dysfunction is a hallmark of aging, but its causes are debated. We show that, during aging, there is a specific loss of mitochondrial, but not nuclear, encoded OXPHOS subunits. We trace the cause to an alternate PGC-1α/β-independent pathway of nuclear-mitochondrial communication that is induced by a decline in nuclear NAD+ and the accumulation of HIF-1α under normoxic conditions, with parallels to Warburg reprogramming. Deleting SIRT1 accelerates this process, whereas raising NAD+ levels in old mice restores mitochondrial function to that of a young mouse in a SIRT1-dependent manner. Thus, a pseudohypoxic state that disrupts PGC-1α/β-independent nuclear-mitochondrial communication contributes to the decline in mitochondrial function with age, a process that is apparently reversible.
Communication breakdown
Mitochondria are often referred to as the cell’s “powerhouse,” generating chemical energy to carry out essential biological functions. These self-contained organelles, which live inside our cells and house their own small genomes, have long been identified as key biological players in aging. As they become increasingly dysfunctional overtime, many age-related conditions such as Alzheimer’s disease and diabetes gradually set in.
Researchers have generally been skeptical of the idea that aging can be reversed, due mainly to the prevailing theory that age-related ills are the result of mutations in mitochondrial DNA—and mutations cannot be reversed.
Sinclair and his group have been studying the fundamental science of aging—which is broadly defined as the gradual decline in function with time—for many years, primarily focusing on a group of genes called sirtuins. Previous studies from his lab showed that one of these genes, SIRT1, was activated by the compound resveratrol, which is found in grapes, red wine and certain nuts.
Ana Gomes, a postdoctoral scientist in the Sinclair lab, had been studying mice in which this SIRT1 gene had been removed. While they accurately predicted that these mice would show signs of aging, including mitochondrial dysfunction, the researchers were surprised to find that most mitochondrial proteins coming from the cell’s nucleus were at normal levels; only those encoded by the mitochondrial genome were reduced.
“This was at odds with what the literature suggested,” said Gomes.
As Gomes and her colleagues investigated potential causes for this, they discovered an intricate cascade of events that begins with a chemical called NAD and concludes with a key molecule that shuttles information and coordinates activities between the cell’s nuclear genome and the mitochondrial genome. Cells stay healthy as long as coordination between the genomes remains fluid. SIRT1’s role is intermediary, akin to a security guard; it assures that a meddlesome molecule called HIF-1 does not interfere with communication.
For reasons still unclear, as we age, levels of the initial chemical NAD decline. Without sufficient NAD, SIRT1 loses its ability to keep tabs on HIF-1. Levels of HIF-1 escalate and begin wreaking havoc on the otherwise smooth cross-genome communication. Over time, the research team found, this loss of communication reduces the cell’s ability to make energy, and signs of aging and disease become apparent.
“This particular component of the aging process had never before been described,” said Gomes.
While the breakdown of this process causes a rapid decline in mitochondrial function, other signs of aging take longer to occur. Gomes found that by administering an endogenous compound that cells transform into NAD, she could repair the broken network and rapidly restore communication and mitochondrial function. If the compound was given early enough—prior to excessive mutation accumulation—within days, some aspects of the aging process could be reversed.
Cancer connection
Examining muscle from two-year-old mice that had been given the NAD-producing compound for just one week, the researchers looked for indicators of insulin resistance, inflammation and muscle wasting. In all three instances, tissue from the mice resembled that of six-month-old mice. In human years, this would be like a 60-year-old converting to a 20-year-old in these specific areas.
One particularly important aspect of this finding involvesHIF-1. More than just an intrusive molecule that foils communication, HIF-1 normally switches on when the body is deprived of oxygen. Otherwise, it remains silent. Cancer, however, is known to activate and hijack HIF-1. Researchers have been investigating the precise role HIF-1 plays in cancer growth.
“It’s certainly significant to find that a molecule that switches on in many cancers also switches on during aging,” said Gomes. “We’re starting to see now that the physiology of cancer is in certain ways similar to the physiology of aging. Perhaps this can explain why the greatest risk of cancer is age.”
“There’s clearly much more work to be done here, but if these results stand, then certain aspects of aging may be reversible if caught early,” said Sinclair.
The researchers are now looking at the longer-term outcomes of the NAD-producing compound in mice and how it affects the mouse as a whole. They are also exploring whether the compound can be used to safely treat rare mitochondrial diseases or more common diseases such as Type 1 and Type 2 diabetes. Longer term, Sinclair plans to test if the compound will give mice a healthier, longer life.
If you liked this article, please give it a quick review on ycombinator or StumbleUpon. Thanks

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.