

There is NO genomic “dark matter” or “missing heritability” — it’s merely a matter of sample size (statistical power) to identify the specific variants that account for the total expected heritability. The paper below (see also HaploSNPs and missing heritability) suggests that essentially all of the expected heritability can be accounted for once rare (MAF less than 0.01) and common SNPs are taken into account. Stephen Hsu suspects the small remaining gap in heritability is accounted for by nonlinear effects.
Researchers propose a method (GREML-LDMS) to estimate heritability for human complex traits in unrelated individuals using whole-genome sequencing data. We demonstrate using simulations based on whole-genome sequencing data that ~97% and ~68% of variation at common and rare variants, respectively, can be captured by imputation. Using the GREML-LDMS method, we estimate from 44,126 unrelated individuals that all ~17 million imputed variants explain 56% (standard error (s.e.) = 2.3%) of variance for height and 27% (s.e. = 2.5%) of variance for body mass index (BMI), and we find evidence that height- and BMI-associated variants have been under natural selection. Considering the imperfect tagging of imputation and potential overestimation of heritability from previous family-based studies, heritability is likely to be 60–70% for height and 30–40% for BMI. Therefore, the missing heritability is small for both traits. For further discovery of genes associated with complex traits, a study design with SNP arrays followed by imputation is more cost-effective than whole-genome sequencing at current prices.
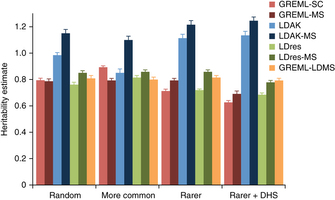
Estimates of heritability using sequence variants under different simulation scenarios based on the UK10K-WGS data set.
SOURCE – Stephen Hsu at Infoproc, Nature Genetics

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.