

A French teenager’s sickle cell disease has been reversed using a pioneering gene therapy treatment. The world-first procedure at Necker Children’s Hospital in Paris offers hope to millions of people with the blood disorder.
Doctors removed his bone marrow – the part of the body that makes blood. They then genetically altered it in a lab to compensate for the defect in his DNA that caused the disease. The altered bone marrow has been making healthy red blood cells for 15 months. He is no longer on any medication.
The doctors are reluctant to call it a cure, but the sickle cell disease is in full remission.
Sickle cell disease causes normally round red blood cells, which carry oxygen around the body, to become shaped like a sickle.
These deformed cells can lock together to block the flow of blood around the body. This can cause intense pain, organ damage and can be fatal.
The teenager who received the treatment had so much internal damage he needed to have his spleen removed and his hips replaced.
Every month he had to go into hospital to have a blood transfusion to dilute his defective blood.
Sickle cell disease (SCD) affects millions of people throughout the world and is particularly common among those whose ancestors came from sub-Saharan Africa; Spanish-speaking regions in the Western Hemisphere (South America, the Caribbean, and Central America); Saudi Arabia; India; and Mediterranean countries such as Turkey, Greece, and Italy.
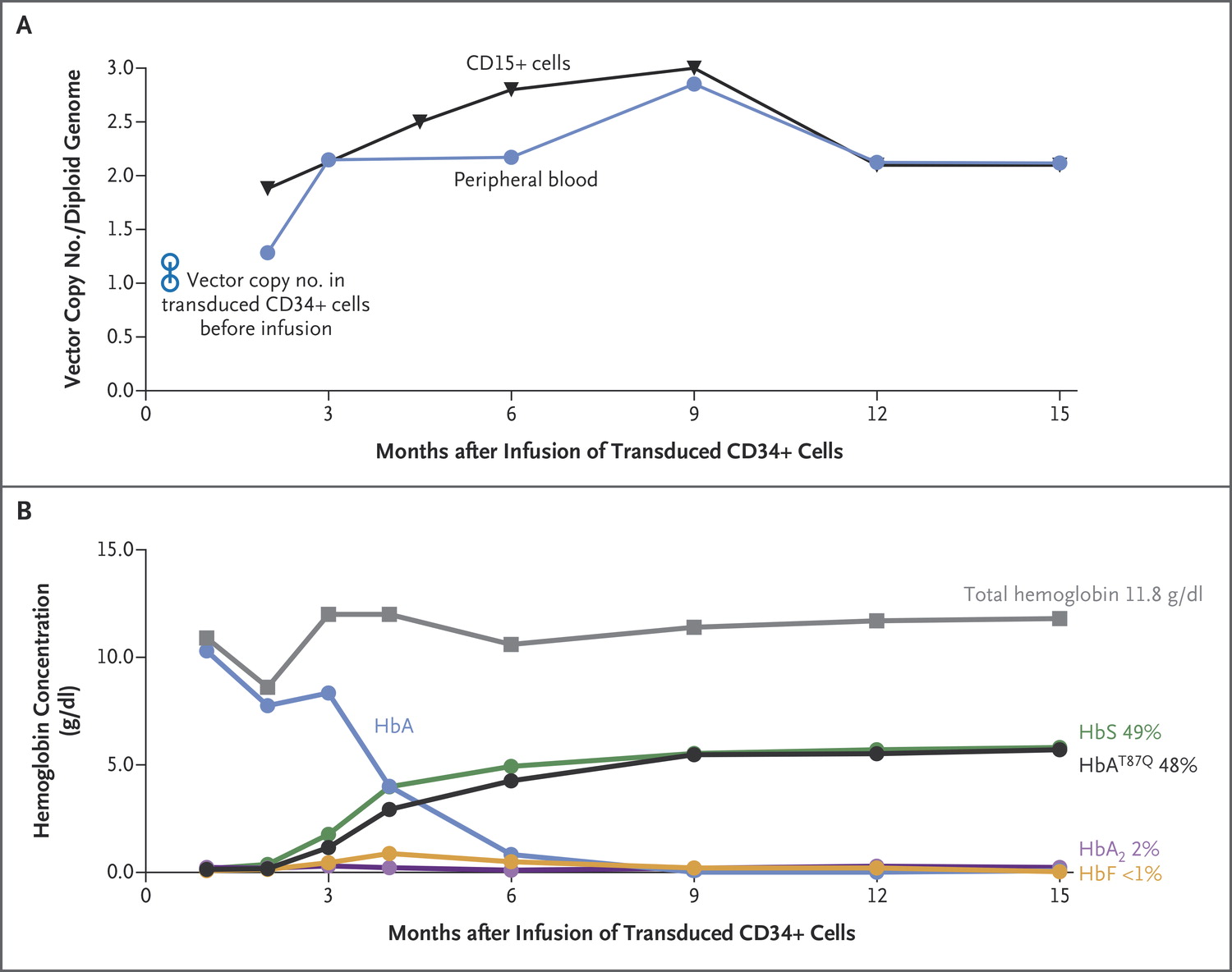
Engraftment with Transduced Cells and Therapeutic Gene Expression in the Patient. Panel A shows vector copy number values in blood nucleated cells and the short-lived CD15+ (neutrophils) fraction thereof over 15 months after infusion of transduced CD34+ cells. Initial values in transduced cells before the infusion are shown. Panel B shows total hemoglobin levels and calculated levels of each hemoglobin fraction based on high-performance liquid chromatography measurements of globin chains. The percent contribution of hemoglobin fractions at month 15 is also indicated. The hemoglobin A (HbA) levels are derived from the regular red-cell transfusions received by the patient before gene therapy and briefly thereafter


New England Journal of Medicine – Gene Therapy in a Patient with Sickle Cell Disease
Abstract
Sickle cell disease results from a homozygous missense mutation in the β-globin gene that causes polymerization of hemoglobin S. Gene therapy for patients with this disorder is complicated by the complex cellular abnormalities and challenges in achieving effective, persistent inhibition of polymerization of hemoglobin S. We describe our first patient treated with lentiviral vector–mediated addition of an antisickling β-globin gene into autologous hematopoietic stem cells. Adverse events were consistent with busulfan conditioning. Fifteen months after treatment, the level of therapeutic antisickling β-globin remained high (approximately 50% of β-like–globin chains) without recurrence of sickle crises and with correction of the biologic hallmarks of the disease. (Funded by Bluebird Bio and others; HGB-205 ClinicalTrials.gov number.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.