
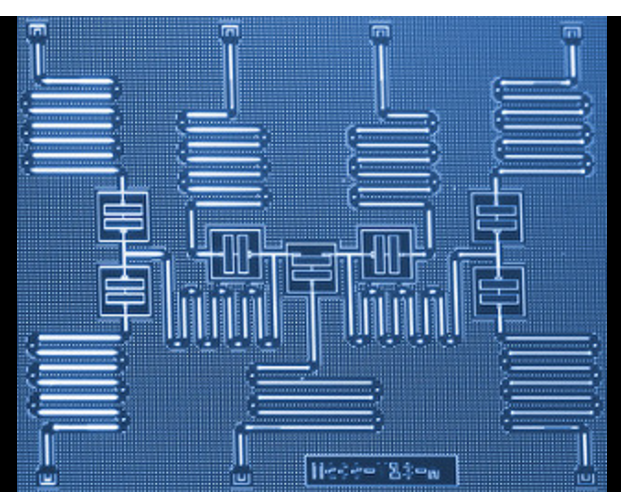
IBM scientists have developed a new approach to simulate molecules on a quantum computer that may one day help revolutionize chemistry and materials science. The scientists successfully used a seven-qubit quantum processor to address the molecular structure problem for beryllium hydride (BeH2) – the largest molecule simulated on a quantum computer to date. The results demonstrate a path of exploration for near-term quantum systems to enhance our understanding of complex chemical reactions that could lead to practical applications.
The team implemented a novel algorithm that is efficient with respect to the number of quantum operations required for the simulation. Using six qubits of a seven-qubit processor they were able to measure BeH2’s lowest energy state, a key measurement for understanding chemical reactions. While this model of BeH2 can be simulated on a classical computer, IBM’s approach has the potential to scale towards investigating larger molecules that would traditionally be seen to be beyond the scope of classical computational methods, as more powerful quantum systems get built. The results were published today as the cover of the peer-reviewed journal Nature.
To help showcase how quantum computers are adept to simulating molecules, developers and users of the IBM Q experience are now able to access a quantum chemistry Jupyter Notebook. The open source quantum chemistry Jupyter Notebook (available through the open access QISKit github repo) allows users to explore a method of ground state energy simulation for small molecules such as hydrogen and lithium hydride. Over a year ago, IBM launched the IBM Q experience by placing a robust five-qubit quantum computer on the cloud for anyone to freely access, and most recently upgraded to a 16-qubit processor available for beta access.
Nature – Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets
The interplay of atoms and molecules is responsible for all matter that surrounds us in the world. However, even today’s most powerful supercomputers cannot exactly simulate the interacting behavior of all the electrons contained in a simple chemical compound such as caffeine. The goal is that we will have the ability to use quantum computers to wholly analyze molecules and chemical reactions, which could help accelerate research and lead to the creation of novel materials, development of more personalized drugs, or discovery of more efficient and sustainable energy sources.
“Thanks to Nobel laureate Richard Feynman, if the public knows one thing about quantum, it knows that nature is quantum mechanical. This is what our latest research is proving – we have the potential to use quantum computers to boost our knowledge of natural phenomena in the world,” said Dario Gil, vice president of AI research and IBM Q, IBM Research. “Over the next few years, we anticipate IBM Q systems’ capabilities to surpass what today’s conventional computers can do, and start becoming a tool for experts in areas such as chemistry, biology, healthcare and materials science.”
“The IBM team carried out an impressive series of experiments that holds the record as the largest molecule ever simulated on a quantum computer,” said Alán Aspuru-Guzik, professor of chemistry and chemical biology at Harvard University. “When quantum computers are able to carry out chemical simulations in a numerically exact way, most likely when we have error correction in place and a large number of logical qubits, the field will be disrupted. Exact predictions will result in molecular design that does not need calibration with experiment. This may lead to the discovery of new small-molecule drugs or organic materials.”
Instead of forcing previously known classical computing methods onto quantum hardware, the scientists reversed the approach by building an algorithm suited to the capability of the current available quantum devices. This allows for extracting the maximal quantum computational power to solve problems that grow exponentially more difficult for classical computers. To characterize the computational power, IBM has adopted a new metric, Quantum Volume. It accounts for the number and quality of qubits, circuit connectivity, and error rates of operations.
Chemistry is one example of a broader set of problems that quantum computers are potentially well-suited to tackle. Quantum computers also have the potential to explore complex optimization routines, as might be found in transportation, logistics or financial services. They could even help advance machine learning and artificial intelligence, which relies on optimization algorithms. Earlier this year, IBM scientists and collaborators demonstrated there is a defined advantage to run a certain type of machine learning algorithm on a quantum computer.
For future quantum applications, IBM anticipates certain parts of a problem to be run on a classical machine while the most computationally difficult tasks might be off-loaded to the quantum computer. This is how businesses and industries will be able to adopt quantum computing into their technology infrastructure and solutions. To get started today, developers, programmers and researchers can run quantum algorithms, work with individual quantum bits, and explore tutorials and simulations on the IBM Q experience. As well, IBM has commercial partners exploring practical quantum applications through the IBM Research Frontiers Institute.
Abstract
Quantum computers can be used to address electronic-structure problems and problems in materials science and condensed matter physics that can be formulated as interacting fermionic problems, problems which stretch the limits of existing high-performance computers. Finding exact solutions to such problems numerically has a computational cost that scales exponentially with the size of the system, and Monte Carlo methods are unsuitable owing to the fermionic sign problem. These limitations of classical computational methods have made solving even few-atom electronic-structure problems interesting for implementation using medium-sized quantum computers. Yet experimental implementations have so far been restricted to molecules involving only hydrogen and helium. Here we demonstrate the experimental optimization of Hamiltonian problems with up to six qubits and more than one hundred Pauli terms, determining the ground-state energy for molecules of increasing size, up to BeH2. We achieve this result by using a variational quantum eigenvalue solver (eigensolver) with efficiently prepared trial states that are tailored specifically to the interactions that are available in our quantum processor, combined with a compact encoding of fermionic Hamiltonians9 and a robust stochastic optimization routine. We demonstrate the flexibility of our approach by applying it to a problem of quantum magnetism, an antiferromagnetic Heisenberg model in an external magnetic field. In all cases, we find agreement between our experiments and numerical simulations using a model of the device with noise. Our results help to elucidate the requirements for scaling the method to larger systems and for bridging the gap between key problems in high-performance computing and their implementation on quantum hardware.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.