
Researchers deliver CRISPR-Cas9 directly into the inner ear hair cells of mice, preventing hearing loss in animal model of genetic progressive deafness.
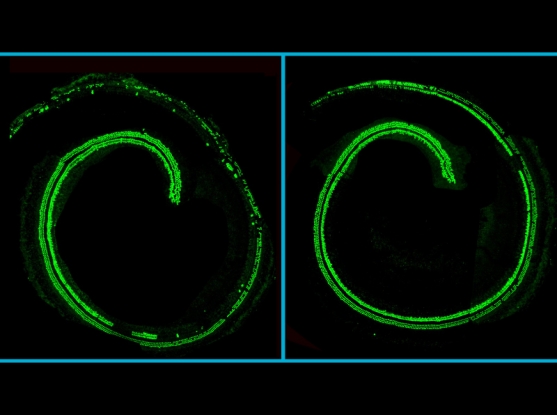
Above – Credit : Gao et al./Nature 2017. Confocal microscopy images of mouse cochlea; hair cells in green. (Left) An untreated cochlea from a mouse with the Tmc1 mutation, displaying hair cell loss. (Right) The opposite, treated, cochlea from the same mouse, displaying hair cell preservation.
Hearing loss is the most common form of sensory loss in humans, and almost half of cases have an underlying genetic cause. Reporting today in Nature, a team led by researchers from the Broad Institute of MIT and Harvard, Massachusetts Eye and Ear, Harvard University, and Howard Hughes Medical Institute (HHMI) have developed a CRISPR-Cas9 genome editing therapy to prevent hearing loss in a mouse model of human genetic progressive deafness.
The therapy delivers a CRISPR-Cas9 gene-editing protein complex directly into the sound-sensing cells of the inner ear (known as “hair cells”) to disrupt a mutation that would otherwise cause the cells to die. The work represents the first time that a genome-editing protein has been ferried directly into the relevant cells to halt progression of genetic hearing loss. Delivering the Cas9 protein itself locally, instead of DNA elements that the cell can use to build Cas9, improved the DNA specificity and potential safety of the treatment.
“We set out to develop a genome-editing strategy to try to address this genetic hearing loss by disrupting the underlying genetic variant,” said co-senior author David Liu, the Richard Merkin Professor, director of the Merkin Institute of Transformative Technologies in Healthcare, and core institute member at the Broad Institute, professor of chemistry and chemical biology at Harvard University, and HHMI investigator. “A lot of additional work is needed before this strategy might inform the development of a therapy for humans, but at this stage, we’re delighted and excited that the treatment preserved some hearing in the animal model.”
Hair cells are the specialized inner-ear cells that turn the mechanical vibrations of sound waves into electrical signals that the brain can interpret. One root cause of genetic hearing loss is a single-letter mutation in a gene called TMC1 that causes hair cells to produce a malformed, toxic protein, one which builds up and kills the cell. Humans (and mice) with this mutation suffer progressive hearing loss during youth, and eventually become profoundly deaf.
Because the mutated TMC1 gene only differs from its normal counterpart by a single letter of DNA, Cas9 must target the mutated gene with laser precision. Otherwise, the Cas9 protein would easily cut and disable the functional copy of the gene instead.
With an eye towards improving the editing precision, the team built on prior work they had published in 2014. When it comes to using CRISPR-Cas9 for gene editing, researchers typically insert the DNA encoding the Cas9 complex into a cell, and let the cell use its own machinery to produce the gene-editing arsenal. But Liu and colleagues previously demonstrated that if the Cas9 gene-editing complex itself was delivered directly into a cell, packaged inside an envelope of lipids, the editing was much more precise. Cas9 itself degrades quickly, and generally cuts its exact target first before moving on to edit similar “off-target” DNA strands — so, when delivered as a protein complex, it usually breaks down before it has a chance to make those mistakes.
“The strategy we used was particularly efficient in targeting dominant genetic hearing loss,” said co-senior author Zheng-Yi Chen, associate professor at Massachusetts Eye and Ear. “In humans, dominant hearing loss generally manifests as late-onset and progressive, therefore providing us with a precious time window for intervention. The therapeutic effect through local inner ear delivery also presents a major advantage in reducing potential risks.”
The researchers tested the method in a mouse model of progressive hearing loss with a mutated Tmc1 gene. Left untreated, the mice experience hearing loss by four weeks of age and profound deafness at eight. The team injected the gene-editing mix into the cochlea of newborn mice genetically destined for profound hearing loss.
The treated mice maintained a substantial amount of their hearing compared to the untreated mice. At four weeks, the untreated mice had a measurable response in their brainstem to sound starting at roughly 80 decibels, the volume of a garbage disposal or a loud radio. But the treated mice responded to sound starting around 65 decibels — approximately the same volume as a typical spoken conversation.
Physiological measurements showed that the hair cells survived at a higher rate in the treated cochlea; genetic sequencing showed that among the edited cells, the mutated copy of Tmc1 had successfully been disrupted 94 percent of the time, and the wild-type allele had only been hit 6 percent of the time. At eight weeks, treated mice also retained their instinctive physical “startle” response to sudden loud sound, while the untreated mice did not respond.
“This is an exciting study that demonstrates the feasibility of a DNA-free, virus-free genome editing strategy for a type of autosomal dominant hearing loss characterized by progressive hair cell loss,” said Tina Stankovic, an associate professor at Mass. Eye and Ear who was not involved with the study. “Augmenting the toolbox to treat genetic deafness is of major significance.”
The team plans to develop the therapy in larger animal models of genetic progressive hearing loss. “These results inform the potential development of a treatment for a subtype of genetic hearing loss, but making sure the method is safe and effective is critically important before we propose moving closer to human trials,” said Liu. “We also recognize the importance and remain mindful of cultural considerations within the Deaf community as this work moves forward.”
Nature – Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents
Although genetic factors contribute to almost half of all cases of deafness, treatment options for genetic deafness are limited. We developed a genome-editing approach to target a dominantly inherited form of genetic deafness. Here we show that cationic lipid-mediated in vivo delivery of Cas9–guide RNA complexes can ameliorate hearing loss in a mouse model of human genetic deafness. We designed and validated, both in vitro and in primary fibroblasts, genome editing agents that preferentially disrupt the dominant deafness-associated allele in the Tmc1 (transmembrane channel-like gene family 1) Beethoven (Bth) mouse model, even though the mutant Tmc1Bth allele differs from the wild-type allele at only a single base pair. Injection of Cas9–guide RNA–lipid complexes targeting the Tmc1Bth allele into the cochlea of neonatal Tmc1Bth/+ mice substantially reduced progressive hearing loss. We observed higher hair cell survival rates and lower auditory brainstem response thresholds in injected ears than in uninjected ears or ears injected with control complexes that targeted an unrelated gene. Enhanced acoustic startle responses were observed among injected compared to uninjected Tmc1Bth/+ mice. These findings suggest that protein–RNA complex delivery of target gene-disrupting agents in vivo is a potential strategy for the treatment of some types of autosomal-dominant hearing loss.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.