

You may have thought the human genome was “solved” decades ago when it was first declared fully sequenced in 2003. However, the “complete” sequencing was incomplete with many gaps and errors and we did not know what almost all of the genes did. 8% of the genome was completely sequenced, due to highly repetitive DNA segments that are difficult to match with the rest of the genome
Now a first truly complete sequencing of the human genome was achieved. The sequencing and analysis were performed by a team of more than 100 people, the so-called Telemere-to-Telomere Consortium, or T2T, named for the telomeres that cap the ends of all chromosomes. The consortium’s gapless version of all 22 autosomes and the X sex chromosome is composed of 3.055 billion base pairs, the units from which chromosomes and our genes are built, and 19,969 protein-coding genes. Of the protein-coding genes, the T2T team found about 2,000 new ones, most of them disabled, but 115 of which may still be expressed. They also found about 2 million additional variants in the human genome, 622 of which occur in medically relevant genes.
Large Scale Gene function Exploration
Seperately, new techniques have been applied to determine the functions of the genes on a genome scale.
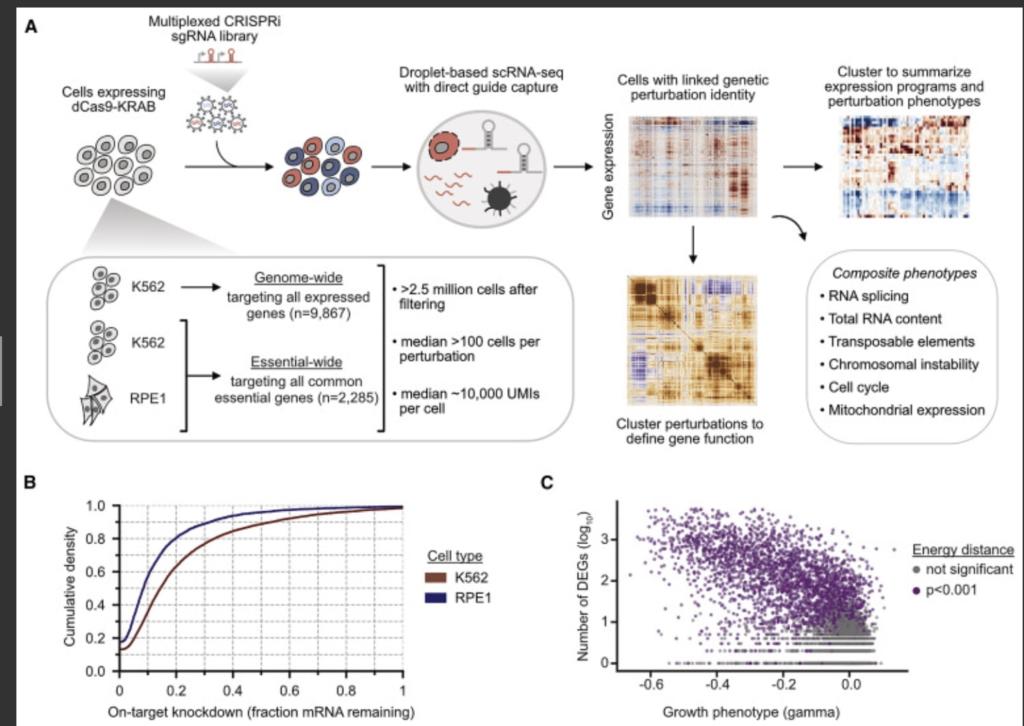
The Perturb-seq method uses CRISPR-Cas9 genome editing to create genetic changes into cells, and then uses single-cell RNA sequencing to capture information about the RNAs that are expressed resulting from a given genetic change. Because RNAs control all aspects of how cells behave, this method can help decode the many cellular effects of genetic changes.
Weissman’s lab and others scaled up the method to the entire genome. They used human blood cancer cell lines as well noncancerous cells derived from the retina. They performed Perturb-seq across more than 2.5 million cells, and used the data to build a comprehensive map tying genotypes to phenotypes.
They looked into genes with unknown functions. They looked at phenotypes of many known genes and compared unknown genes to known ones and look for similar transcriptional outcomes. This could identify genes working together as part of a larger complex.
The mutation of one gene called C7orf26 in particular stood out. the researchers were able to confirm that C7orf26 made up a 15th component of a previously known 14-component complex.
Cell Journal- Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq
Highlights
• Perturb-seq maps the transcriptional effects of genetic perturbations at genome scale
• Transcriptional signatures allow prediction of function for thousands of genes
• Single-cell RNA-seq enables the study of complex composite phenotypes like aneuploidy
• Analysis of mitochondrial genome expression reveals diverse, stress-specific regulation
Summary
A central goal of genetics is to define the relationships between genotypes and phenotypes. High-content phenotypic screens such as Perturb-seq (CRISPR-based screens with single-cell RNA-sequencing readouts) enable massively parallel functional genomic mapping but, to date, have been used at limited scales. Here, we perform genome-scale Perturb-seq targeting all expressed genes with CRISPR interference (CRISPRi) across >2.5 million human cells. We use transcriptional phenotypes to predict the function of poorly characterized genes, uncovering new regulators of ribosome biogenesis (including CCDC86, ZNF236, and SPATA5L1), transcription (C7orf26), and mitochondrial respiration (TMEM242). In addition to assigning gene function, single-cell transcriptional phenotypes allow for in-depth dissection of complex cellular phenomena—from RNA processing to differentiation. We leverage this ability to systematically identify genetic drivers and consequences of aneuploidy and to discover an unanticipated layer of stress-specific regulation of the mitochondrial genome. Our information-rich genotype-phenotype map reveals a multidimensional portrait of gene and cellular function.
The study presents a blueprint for the construction and analysis of rich genotype-phenotype maps to serve as a driving force for the systematic exploration of genetic and cellular function.
SOURCE – Journal Cell
Written By Brian Wang, Nextbigfuture.com

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.
Hopefully, in my lifetime medical science will figure out a way to “remove” or “unplug” the Diabetes gene.
Obviously, we have no idea of what unintended consequences might occur, but to begin that group of experiments would certainly be exciting.
Observationally we already know the way to keep genes that produce type 2 diabetes from causing the condition. Restrict eating excessive carbohydrates. Type 2 diabetes pretty much only occurs in populations that chronically eat a lot more carbs than ancestral populations ever would have. There is a lot of individual difference in tolerance basically in how much insulin your pancreas can produce before it’s disregulated but the pattern is very simple.
He is probably referring to Type 1 Diabetics. They have identified a few genes in this area. They can actually tell a child a year or two out they will most likely be type 1 and need insulin using a blood test. Now if we can turn those genes off…
There may be genes that increase one’s vulnerability to type II diabetes, but any that “cause” it, would be held to less than 1% of the population. That is just the way genetics works.
I think the real cause is the body’s reaction to the superantigen of Staphylococcus: https://now.uiowa.edu/2015/06/bacteria-may-cause-type-2-diabetes
“as people gain weight, they are increasingly likely to be colonized by staph bacteria”. My hypothesis is that it leads to skin folds and incomplete cleaning when bathing/showering that increase the numbers of staph bacteria.
In any case, weight loss probably helps. There is a very impressive weight loss drug that was just approved in the US and marketed as Wegovy. Sadly, the manufacturers are incredibly greedy.
We may know a smidgen more that we did in 2003, but not much. The most common genetic deletion is called 22q11.2 DS. The deletion causes up to 200 different health symptoms, along with a 20% chance of schizophrenia. Within that genetic address (22q11.2), they are still a long way from piecing together what DNA within that address causes what problems.