

Harvard researchers bring the accuracy, sample efficiency, and robustness of deep equivariant neural networks to the simulate 44 million atoms. This is achieved through a combination of innovative model architecture, massive parallelization, and models and implementations optimized for efficient GPU utilization. The resulting Allegro architecture bridges the accuracy speed tradeoff of atomistic simulations and enables description of dynamics in structures of unprecedented complexity at quantum fidelity. To illustrate the scalability of Allegro, they perform nanoseconds-long stable simulations of protein dynamics and scale up to a 44-million atom structure of a complete, all-atom, explicitly solvated HIV capsid on the Perlmutter supercomputer. They demonstrate excellent strong scaling up to 100 million atoms and 70% weak scaling to 5120 A100 GPUs.
This is the first scalable, transferable machine-learning potential with state-of-the-art equivariant deep-learning accuracy. Performance of 100 timesteps/s for range of biomolecular systems. 70% weak scaling to 1280 nodes and 5120 A100 GPUs, excellent strong scaling up to 100 million atoms. First application of state-of-the-art machine learning interatomic potentials to large-scale biomolecular simulations.

Problem Overview: First-Principles Dynamics of Matter
The ability to predict the time evolution of matter on the atomic scale is the foundation of modern computational biology, chemistry, and materials engineering. Even as quantum mechanics governs the microscopic atom-electron interactions in vibrations, migration and bond dissociation, phenomena governing observable physical and chemical processes often occur at much larger length- and longer time-scales than
those of atomic motion. Bridging these scales requires both innovation in fast and highly accurate computational approaches capturing the quantum interactions and in extremely parallelizable architectures able to access exascale computers.
Presently, realistic physical and chemical systems are far more structurally complex than what computational methods are capable of investigating, and their observable evolution is beyond the timescales of atomistic simulations. This gap between key fundamental questions and phenomena that can be effectively modeled has persisted for decades. From one side of the gap, models of small size, representing ostensibly important parts of the systems, can be constructed and investigated with highfidelity computationally expensive models, such as electronic structure methods of density functional theory (DFT) and wave-function quantum chemistry. In the domain of materials science, these models can capture individual interfaces in metallic composites, defects in semiconductors, and flat surfaces of catalysts. However, evolution of such structures over relevant time scales is out of reach with electronic structure methods. Importantly, such reduction of complexity
is not possible in the domain of biological sciences, where entire structures of viruses consist of millions of atoms, in addition to similarly large number of explicit water molecules needed to capture the physiological environment. From the other side of the gap, uncontrolled approximations have to be made to reach large sizes and sufficient computational speeds. These approximations have relied on very simple analytical models for interatomic interactions and have many documented failures of describing dynamics of both complex
inorganic and biological materials.
Molecular dynamics (MD) simulations are a pillar of computational science, enabling insights into the dynamics of molecules and materials at the atomic scale. MD provides a level of resolution, understanding, and control that experiments often cannot provide, thereby serving as an extremely powerful tool to advance our unstanding and design of novel molecules and materials. Molecular dynamics simulates the time evolution of atoms according to Newton’s equations of motion. By integrating the forces at each time step, a sequence of many-atom configurations is obtained, from which physical observables can then be obtained.
The bottleneck of MD is the short integration time step required, which usually lies on the order of femtoseconds. Since many chemical and biological processes occur on the timescale of microseconds or even milliseconds, billions to trillions of integration steps are needed. This highlights the requirement common to all MD simulation for access to atomic forces in a way that is simultaneously both a) accurate and b) computationally efficient.
For decades, two different avenues have been pursued: on one hand, classical force-fields (FFs) are able to simulate large-scale systems with
high computational efficiency, enabling simulations of billions of atoms and reaching even microsecond simulation timescales on special-purpose hardware The simple functional form of classical FFs, however, greatly limits their predictive power and results in them not being able to capture quantum mechanical effects or complex chemical transformations. On the other hand, quantum-mechanical simulations provide a
highly accurate description of the electronic structure of molecules and materials. Interatomic forces computed with these first-principles methods can then be used to integrate the atoms in molecular dynamics, called ab-initio molecular dynamics (AIMD), with DFT being the most common method of choice. The cubic scaling of plane-wave DFT with the number of electrons, however, is the key bottleneck limiting AIMD simulations as it severely limits both length- and timescales of AIMD simulations, only allowing thousands of atoms and hundreds of picoseconds in routine simulations.
Current State of the Art: Accuracy
Over the past two decades, machine learning interatomic potentials (MLIPs) have been pursued with immense interest as a third approach, promising to bypass this long-standing dilemma. The aim of MLIPs is to learn energies and forces from high-accuracy reference data
while scaling linearly with the number of atoms. Initial efforts combined hand-crafted descriptors with a Gaussian Process or a shallow neural network. These first MLIPs were quickly further improved and have been adapted to biomolecular simulations, trained on large data sets to
provide general-purpose potentials. Despite this initial progress, however, this first generation of MLIPs has been severely limited in predictive accuracy, often unable to generalize to structures different from those in the training data, resulting in simulations that are not transferable and often not robust. In an effort to overcome these limitations in accuracy, more recently, deep learning interatomic potentials based on the message passing neural network (MPNN) paradigm have been proposed and shown to be a more accurate alternative to the first generation of MLIPs, however at a significant computational overhead.
Common to all interatomic potentials is a focus on symmetry: in particular, the energy must obey invariance with respect to translations, rotations, and reflections, which together comprise E(3), the Euclidean group in 3D. Both classical forcefields and modern MLIPs achieve this by only ever acting on geometric invariants of the underlying structures. More recently, in an attempt to better represent the symmetry of the data, invariant interatomic potentials have been generalized to equivariant ones. In equivariant MLIPs, the hidden network features consist of not only scalar features, but also vectors and higher-order tensors, resulting in a more faithful representation of the atomistic geometry.
The initial NequIP work on equivariant interatomic potentials demonstrated not only a step change in state-of-theart accuracy, but also the ability to outperform the invariant DeepMD accuracy while training on a thousand times smaller data set. Following these efforts, equivariant MLIPs have been shown to greatly improve stability of simulations and display much better extrapolative power than existing approaches. Equivariant potentials however, struggle with scale and speed since all existing methods combine the equivariance with the MPNN architecture of previous generation methods. This is due to the graph propagation mechanism in MPNNs: at each layer of message passing, information is passed from a central node to its neighbors. The best system has achieved a 20,894 atom simulation.
In order to scale a message passing network, one would have to either maintain enormous neighbor lists, or transfer messages and gradients between devices at each layer. Both of these approaches are destined to be slow, with the latter also requiring extensive software development to allow the neural network’s message passing to work together with the MD software’s spatial decomposition message passing. Thus the exceptional accuracy of equivariant methods remains inaccessible for applications that require large length scales and long timescale simulations, with biological systems being a major example that is virtually entirely excluded from these state-of-the-art approaches.
Current State of the Art: Scalability and speed
In parallel to the improvements in accuracy, great strides have been achieved in terms of the computational cost of MLIPs, with several methods sacrificing some accuracy in order to be able to perform extreme-scale simulations. Most notably, the DeePMD method won the 2020 Gordon Bell supercomputing award for their 100 million-atom simulations, using the entire Summit machine.
SNAP extended the scale to 20 billion atoms, accompanied by a 20-fold increase in speed. Finally, FLARE set the current record for GPU-accelerated MLIP uncertainty-aware reactive MD benchmarks with 0.5 trillion atoms on Summit and a 70% speed increase over SNAP.
Allegro: Scalable Equivariant Deep Learning
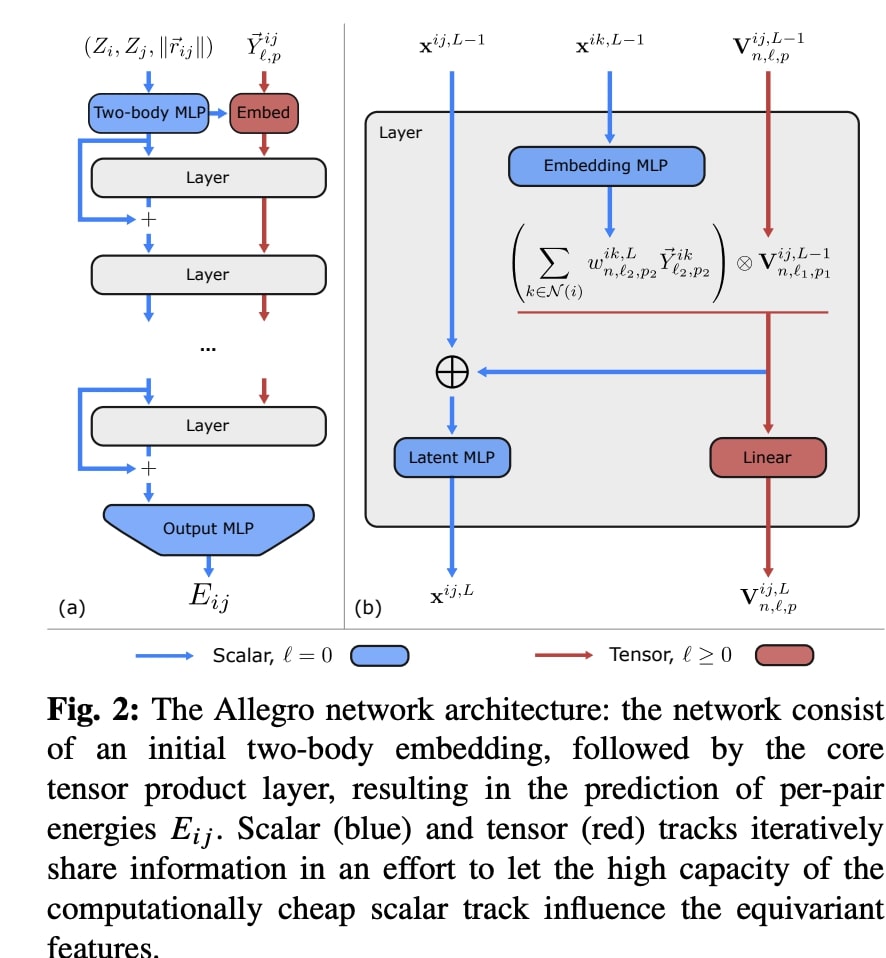
Allegro is the first scalable equivariant MLIP to overcome the previous dilemma of choosing between highly accurate, robust, transferable equivariant models and scalable, but less accurate, first-generation models. Allegro attains this by first decomposing the total predicted energy of the system into atomic energies.
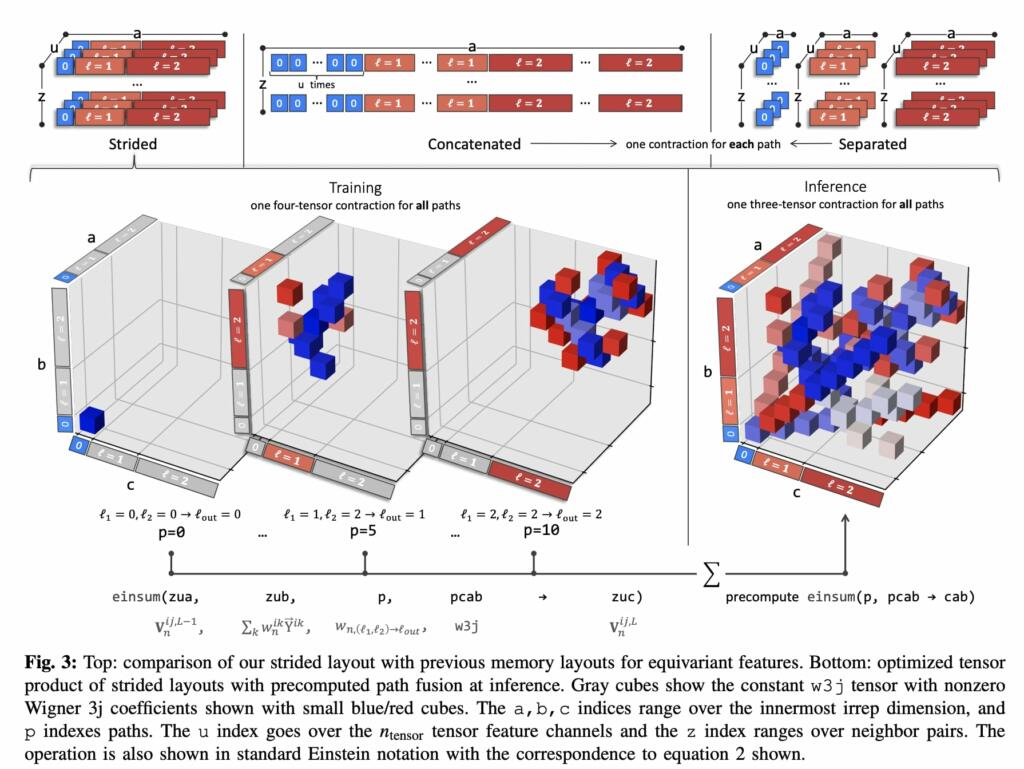
The first key innovation of Allegro is its tensor product layer, which updates the features with information about neighboring atoms’ geometry using the “tensor product of representations,” a fundamental equivariant operation. Specifically, the perordered-pair tensor features are updated by taking their tensor product with a learned weighted sum over the spherical harmonic embeddings of the positions of the neighbors of the central atom.
This innovation allows Allegro to learn increasingly complex representations of the atomic structure layer after layer while keeping all interactions strictly local, thus making it massively parallelizable.
The second central innovation of Allegro is motivated by the difference in cost between scalar operations and the much more expensive 𝑂(3)-equivariant operations that are symmetrically permitted on tensors. Allegro is therefore designed to put significant network capacity into the scalar operations, particularly dense neural networks, which are comparatively cheap and highly optimized on modern GPU hardware, while keeping as few expensive equivariant operations as possible (only the tensor product) and limiting the number of tensor feature channels.
This is achieved by having separate scalar and tensor tracks throughout the network, which at each layer communicate information which allows the high capacity of the scalar track to “control” the equivariant features. Across deep learning, it has repeatedly been observed that larger networks perform better. In high-performance applications of deep learning such as interatomic potentials, however, this increase in computation is unsustainable as it would hurt computational efficiency. The ability of Allegro to partially decouple these two effects allows it to greatly increase network capacity while keeping the computational overhead moderate.
Allegro outperforms state-of-the-art potentials on energies and forces of small molecules as measured by the QM9 and revMD17 benchmarks, including state-of-the-art equivariant message-passing-based approaches. Remarkably, Allegro also performs significantly better than the DeepMD potential on water despite being trained on more than 1,000 times fewer reference data. This demonstrates that Allegro is able to retain the remarkable improvements demonstrated by equivariant message-passing potentials, while being massively parallelizable, greatly increasing accessible length-scales as well as time-to-solution.
The work demonstrates the advantages of high-capacity equivariant Allegro models in accurately learning forces across the entire SPICE dataset of over 1 million structures of drug-like molecules and peptides. This data scale implies the promise of learning the entire sets of inorganic materials and organic molecules far more accurately than previously attempted, which would open the prospects of fast exascale simulations of unprecedentedly wide ranges of materials systems.
Recently they demonstrated that it is possible to efficiently quantify uncertainty of deep equivariant model predictions of forces and energies and use it to perform active learning for automatic construction of training sets. Natural adaptation of Gaussian mixture models in Allegro will open the possibility of large-scale uncertainty-aware simulations using a single model, as opposed to ensembles.
Finally, the major implication of the demonstrated accuracy of equivariant models is the urgent need to improve the accuracy and efficiency of the quantum electron structure calculations that are used as reference to train MLIPs, as this now becomes the major accuracy bottleneck in computational chemistry, biology and materials science.

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.