
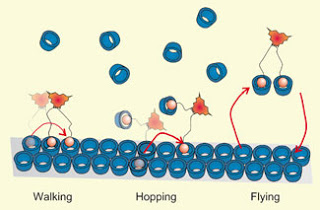

The mechanism of molecular motion changes as the environment changes; in this case, due to competition from free receptors (blue) in solution
Controlling how molecules move on surfaces could be the key to more potent drugs that block the attachment of viruses to cells, and will also speed development of new materials for electronics and energy applications. Tyndall National Institute, UCC researcher Dr. Thompson performed computer simulations that enabled a greater understanding of how two-legged molecules move along patterned surfaces, in a kind of molecular hopscotch.
Widespread industrial uptake of nanotechnology requires cheap, easy and robust solutions that allow manipulation of matter at the smallest scales and so a key enabling feature will be the ability to move material around molecule by molecule. One of the major difficulties is the very different physics that operates at the scale of atoms and molecules; water, for example, feels more like treacle to a molecule, and molecules tend to huddle and stick together due to microscopic forces between their atoms. Dr. Thompson explains: “The experiments performed by the group at Twente were very elegant. They involved making two-legged molecules and using a fluorescence microscope to watch how they move along a wet surface. The molecules are hydrophobic, meaning they don’t like water, and the surface was pockmarked with hydrophobic cavities so a weak glueing interaction, based on a mutual dislike of water, drives the interaction between the molecules and the surface.
While the energetics of this type of interaction was worked out over a decade ago by George Whitesides’s group at Harvard, it’s usefulness for materials development was limited because little was known until now on the paths that the molecules take”.
Because the molecules have multiple legs, they display a surprisingly rich behaviour at the surface, beyond simply attaching/detaching, with Dr. Thompson’s computer simulations complementing the experiments and showing the different mechanisms by which the molecules move. The motion switches from walking to hopping to flying, as the environment changes.
Dr. Thompson continues: “Access to high performance computing facilities enabled us to model the different pathways and aid interpretation of the microscopy results. We ran most of the simulations on our own Science Foundation Ireland-supported computing clusters at Tyndall, and also did a few larger-scale calculations at the Irish Center for High End Computing. It’s an exciting time for research as experiments and simulations are finally on the same page; the experiments can finally drill down far enough to see molecule-scale features while advances in computing mean we can routinely model systems composed of hundreds of thousands, and even millions, of atoms”.
The kinetics of multivalent (multisite) interactions at interfaces is poorly understood, despite its fundamental importance for molecular or biomolecular motion and molecular recognition events at biological interfaces. Here, we use fluorescence microscopy to monitor the spreading of mono-, di- and trivalent ligand molecules on a receptor-functionalized surface, and perform multiscale computer simulations to understand the surface diffusion mechanisms. Analogous to chemotaxis, we found that the spreading is directional (along a developing gradient of vacant receptor sites) and is strongly dependent on ligand valency and concentration of a competing monovalent receptor in solution. We identify multiple surface diffusion mechanisms, which we call walking, hopping and flying. The study shows that the interfacial behaviour of multivalent systems is much more complex than that of monovalent ones.

Representative structure from the molecular dynamics simulations, showing a planar view of the model used to describe the interaction of the GII divalent molecule with the CD-functionalized surface. Water molecules and hydrogen atoms are omitted for clarity. The central orthorhombic unit cell is marked by the black rhombus; expansion with periodic boundary conditions provides the extended hexagonally-packed CD-functionalized surface. The GII molecule is colored blue with Ad anchors shown as spheres and the remaining atoms shown as sticks. The oxygens of the OH groups at the entrance to the CD cavities are shown as red spheres with the remaining atoms shown as brown sticks. The underlying linkers and substrate, arbitrarily set in the simulations to alkanethioether chains and Au(111), are shown as brown sticks and gold spheres. Surface binding sites are labelled in the central unit cell, with Hs1 and Hs2 denoting respectively the CD site binding GII group Ad1 and Ad2. The remaining two uncomplexed surface sites are labelled Hs-free. The snapshot shows the monovalent GII(Hs)1 complex formed after releasing Ad1 from site Hs1.
27 pages of supplemental information
If you liked this article, please give it a quick review on ycombinator or StumbleUpon. Thanks

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.